Research overview
Below are four research directions that are currently of great interest to the group. That said, we are always open to exploring new ideas and collaborations.
1. Elucidating disease biology from genetic association studies
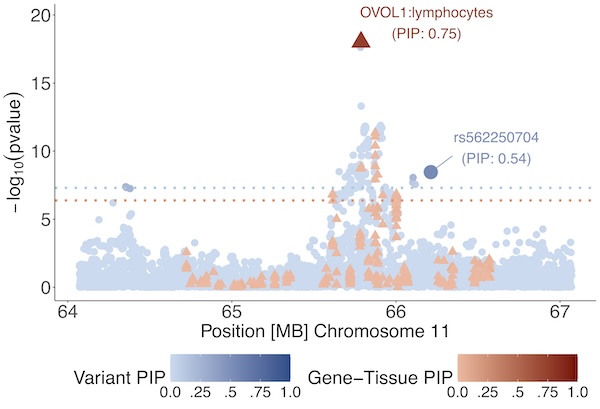
A major goal of the lab is to develop integrative statistical approaches to decode variant-to-gene regulation or disease and infer biological mechanisms underlying disease-associated genetic variants. This work will rely on integration of GWAS data with diverse variant-to-gene linking strategies derived from functional genomic datasets, including QTL analyses, CRISPR-based perturbations, enhancer–gene connections from single-cell multiome assays, and chromatin contact maps.
As an example, we developed Tissue-Gene Fine-Mapping (TGFM), a statistical method to uncover the functional mechanisms underlying disease-associated variants by integrating multi-tissue eQTL data with GWAS data (Strober et al., Nature Genetics 2025). TGFM exploits variation in eQTL effects across tissues to pinpoint both the causal gene and its tissue of action at each GWAS locus. Causal gene-tissue pairs identified by TGFM reflected both known biology (for example, TPO–thyroid for hypothyroidism) and biologically plausible findings (for example, SLC20A2–artery aorta for diastolic blood pressure).
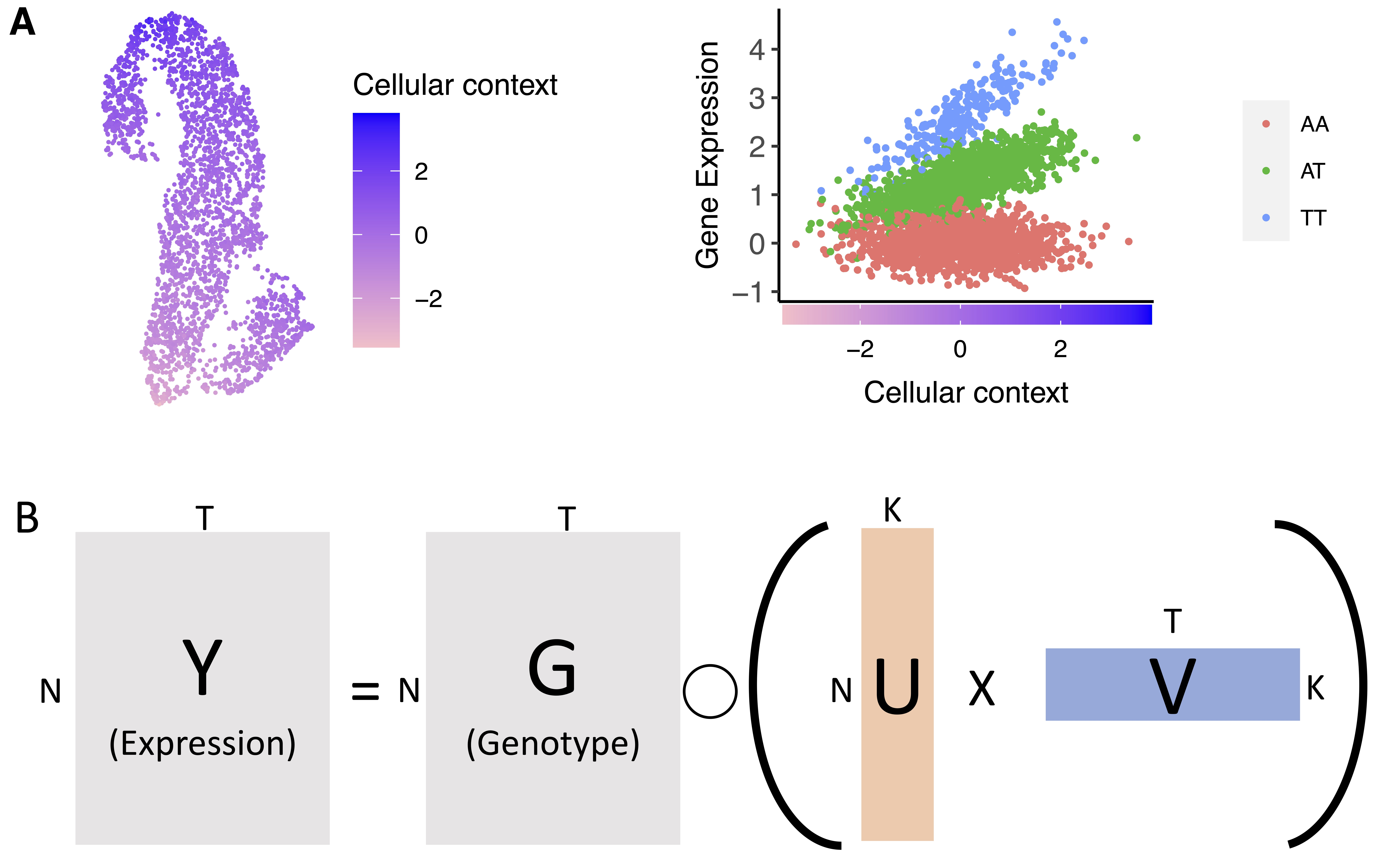
2. Modeling genetic effects on gene expression across tissues, cell-types, and contexts
3. Advancing genomically-informed precision medicine for rare diseases
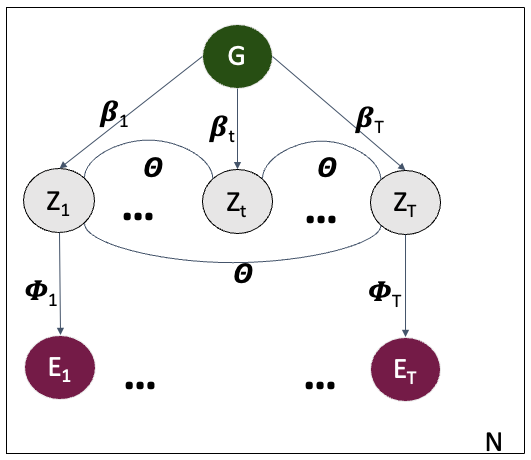
Looking ahead, we are enthusiastic about continuing this line of work and developing new methods to advance precision medicine for rare disease cases at Boston Children's Hospital. Specifically, we aim to build well-calibrated models that predict disease status or treatment response by integrating clinical, genetic, and multi-omic data. By well-calibrated, we mean that when our model predicts a 70% probability of treatment response, approximately 70% of future patients with that prediction will, in fact, respond. We believe that well-calibrated predictions are essential for adoption of genomically-informed precision medicine into practice.
4. Inferring variant-to-gene links using deep-learning sequence-to-expression models
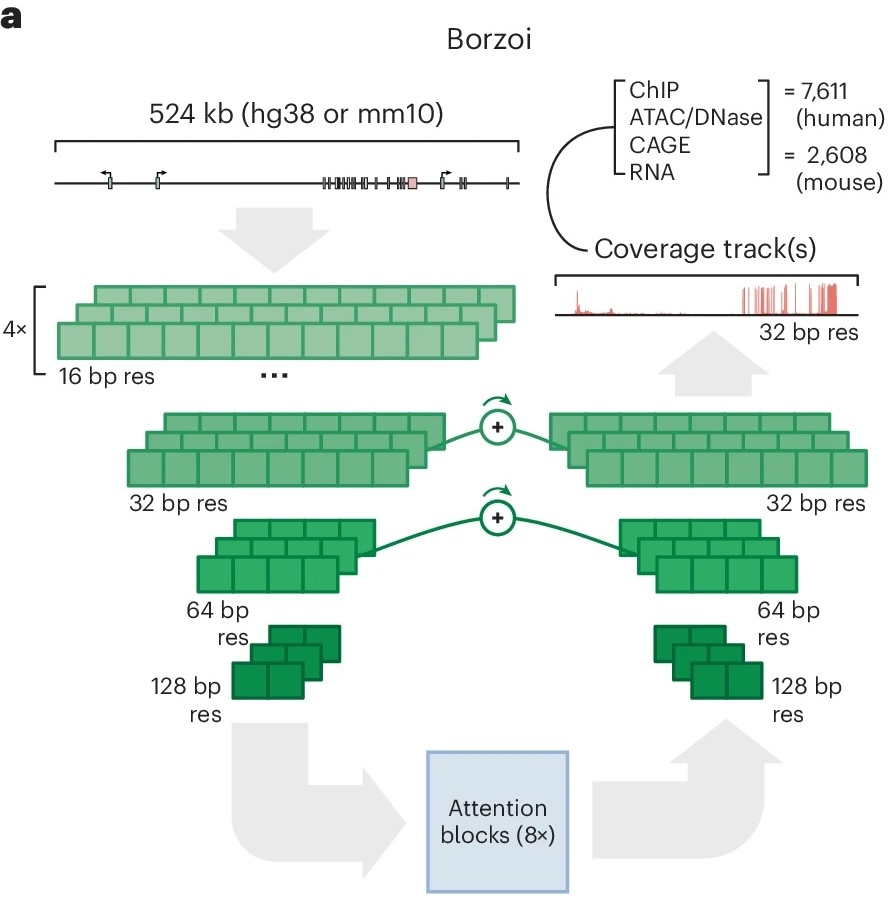
S2E models (e.g., AlphaGenome, Borzoi) leverage deep learning to predict gene expression across tissues, cell types, and contexts directly from DNA sequence. They can also estimate a variant’s effect on a gene by computing the predicted change in expression after substituting the reference with the alternative allele. This provides a complementary alternative to QTL-based estimates. Notably, S2E-predicted effects can be derived without population-scale data, making them particularly valuable for hard-to-assay contexts.
We are currently developing new approaches to integrate S2E predictions with GWAS data to gain insight into the functional mechanisms that underlie disease-associated variants.